
Map gene regulation and expression with spatial context.
Spatial CoProfiling simultaneously measures chromatin accessibility and RNA from the same tissue section — directly linking regulatory state to transcriptional output, pixel by pixel, within intact tissue architecture.
Fan Lab · Yale
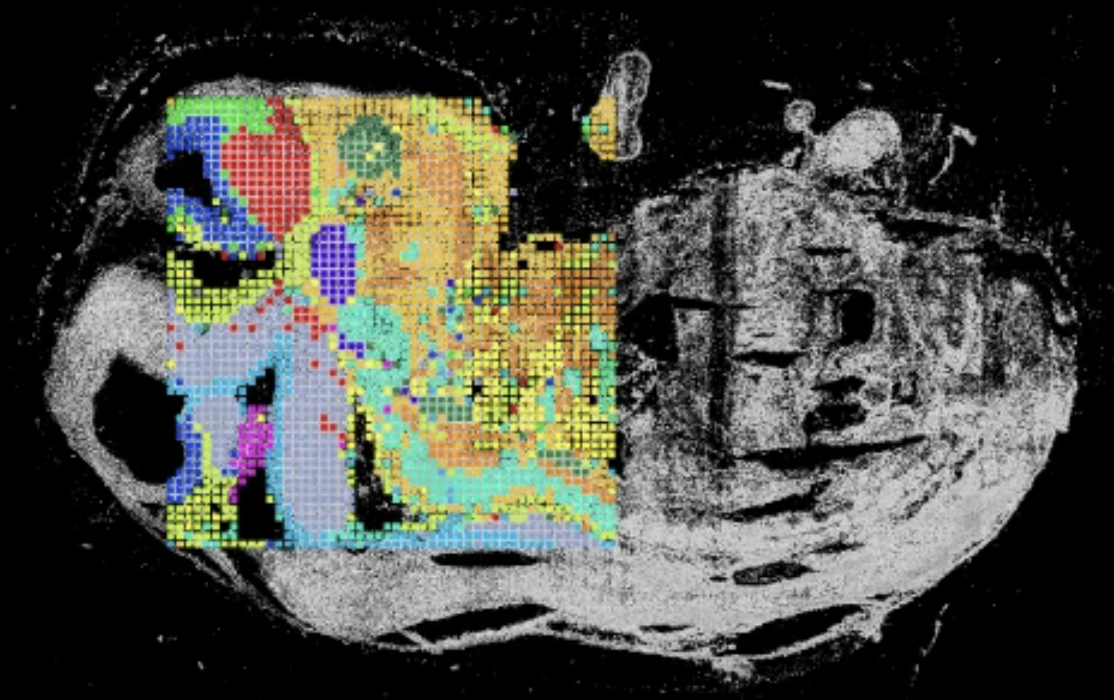
Spatial ATAC–RNA co-profiling from the same section
Built on the spatial ATAC–RNA-seq method first described in a 2023 Nature publication from the Fan Lab at Yale, demonstrating near-single-cell resolution co-profiling of the epigenome and transcriptome in mammalian tissues.
-
Paired ATAC + RNA from the same section Measure chromatin accessibility and gene expression simultaneously — no serial sectioning or data integration assumptions required.
-
Direct regulatory interpretation Link open chromatin peaks to active transcription in the same spatial pixel, revealing regulatory programs driving cell states in context.
-
Spatially resolved chromatin–expression coupling Identify where regulatory priming precedes transcriptional changes across tissue architecture — not possible with bulk or single-cell methods alone.
-
Compatible with downstream epigenomic analysis ATAC data from the same section can be analyzed with standard peak-calling, motif scoring, and TF activity inference pipelines.

White papers and conference posters.
Technical references and application examples for spatial CoProfiling across tissue types and disease contexts.
- Paired ATAC + RNA from the same tissue section
- Direct regulatory interpretation at spatial resolution
- Spatially resolved chromatin–expression coupling
- Open chromatin detected ahead of RNA changes, revealing regulatory priming in select tumor clusters
- Distinct TF motif enrichment between primary breast tumor and liver metastasis
- Integrated ATAC + RNA clustering identifies heterogeneous tumor states
Ready to co-profile your tissue?
Our team will help you qualify your samples and design a CoProfiling experiment that answers your regulatory questions.